Anti-Collagen I antibody (ab233080)
")
Key features and details
- Rabbit polyclonal to Collagen I
- Suitable for: WB, IHC-P
- Reacts with: Human, Pig
- Isotype: IgG
Get better batch-to-batch reproducibility with a recombinant antibody


- Research with confidence – consistent and reproducible results with every batch
- Long-term and scalable supply – powered by recombinant technology for fast production
- Success from the first experiment – confirmed specificity through extensive validation
- Ethical standards compliant – production is animal-free
Overview
-
Product name
Anti-Collagen I antibody
See all Collagen I primary antibodies -
Description
Rabbit polyclonal to Collagen I -
Host species
Rabbit -
Tested applications
Suitable for: WB, IHC-Pmore details -
Species reactivity
Reacts with: Human, Pig
Predicted to work with: Mouse, Rat, Chicken, Cow, Dog
-
Immunogen
Recombinant fragment (His-tag) corresponding to Human Collagen I aa 1200-1450. (Expressed in E.coli).
Database link: P02452 -
Positive control
- IHC-P: Human kidney tissue. WB: Recombinant human Collagen I protein; Human placenta, liver, and Pig skin lysates.
-
General notes
The Life Science industry has been in the grips of a reproducibility crisis for a number of years. Abcam is leading the way in addressing this with our range of recombinant monoclonal antibodies and knockout edited cell lines for gold-standard validation. Please check that this product meets your needs before purchasing.
If you have any questions, special requirements or concerns, please send us an inquiry and/or contact our Support team ahead of purchase. Recommended alternatives for this product can be found below, along with publications, customer reviews and Q&As
Properties
-
Form
Liquid -
Storage instructions
Shipped at 4°C. Store at +4°C short term (1-2 weeks). Upon delivery aliquot. Store at -20°C long term. Avoid freeze / thaw cycle. -
Storage buffer
pH: 7.40
Preservative: 0.011% Proclin 300
Constituents: 55.77% Glycerol (glycerin, glycerine), 44.219% PBS -
 Concentration information loading...
Concentration information loading... -
Purity
Immunogen affinity purified -
Purification notes
ab233080 was purified by antigen-specific affinity chromatography followed by Protein A affinity chromatography. -
Clonality
Polyclonal -
Isotype
IgG -
Research areas
Associated products
-
Compatible Secondaries
-
Isotype control
-
Recombinant Protein
Applications
The Abpromise guarantee
Our Abpromise guarantee covers the use of ab233080 in the following tested applications.
The application notes include recommended starting dilutions; optimal dilutions/concentrations should be determined by the end user.
| Application | Abreviews | Notes |
|---|---|---|
| WB |
Use a concentration of 1 - 5 µg/ml. Predicted molecular weight: 139 kDa.
Positive Control: Hu stomach, skin and adrenal gland tissue lysates. Acid or enzyme treatment with pepsin is a better method to isolate collagen. Continuous refrigeration throughout collagen extraction is important to avoid degradation and denaturation. Take care with pH, temperature, and concentration to avoid collagen polymerization. |
|
| IHC-P |
Use a concentration of 5 - 20 µg/ml.
|
| Notes |
|---|
|
WB
Use a concentration of 1 - 5 µg/ml. Predicted molecular weight: 139 kDa. Positive Control: Hu stomach, skin and adrenal gland tissue lysates. Acid or enzyme treatment with pepsin is a better method to isolate collagen. Continuous refrigeration throughout collagen extraction is important to avoid degradation and denaturation. Take care with pH, temperature, and concentration to avoid collagen polymerization. |
|
IHC-P
Use a concentration of 5 - 20 µg/ml. |
Target
-
Function
Type I collagen is a member of group I collagen (fibrillar forming collagen). -
Tissue specificity
Forms the fibrils of tendon, ligaments and bones. In bones the fibrils are mineralized with calcium hydroxyapatite. -
Involvement in disease
Defects in COL1A1 are the cause of Caffey disease (CAFFD) [MIM:114000]; also known as infantile cortical hyperostosis. Caffey disease is characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age.
Defects in COL1A1 are a cause of Ehlers-Danlos syndrome type 1 (EDS1) [MIM:130000]; also known as Ehlers-Danlos syndrome gravis. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS1 is the severe form of classic Ehlers-Danlos syndrome.
Defects in COL1A1 are the cause of Ehlers-Danlos syndrome type 7A (EDS7A) [MIM:130060]; also known as autosomal dominant Ehlers-Danlos syndrome type VII. EDS is a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDS7A is marked by bilateral congenital hip dislocation, hyperlaxity of the joints, and recurrent partial dislocations.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 1 (OI1) [MIM:166200]. A dominantly inherited connective tissue disorder characterized by bone fragility and blue sclerae. Osteogenesis imperfecta type 1 is non-deforming with normal height or mild short stature, and no dentinogenesis imperfecta.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 2A (OI2A) [MIM:166210]; also known as osteogenesis imperfecta congenita. A connective tissue disorder characterized by bone fragility, with many perinatal fractures, severe bowing of long bones, undermineralization, and death in the perinatal period due to respiratory insufficiency.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 3 (OI3) [MIM:259420]. A connective tissue disorder characterized by progressively deforming bones, very short stature, a triangular face, severe scoliosis, grayish sclera, and dentinogenesis imperfecta.
Defects in COL1A1 are a cause of osteogenesis imperfecta type 4 (OI4) [MIM:166220]; also known as osteogenesis imperfecta with normal sclerae. A connective tissue disorder characterized by moderately short stature, mild to moderate scoliosis, grayish or white sclera and dentinogenesis imperfecta.
Genetic variations in COL1A1 are a cause of susceptibility to osteoporosis (OSTEOP) [MIM:166710]; also known as involutional or senile osteoporosis or postmenopausal osteoporosis. Osteoporosis is characterized by reduced bone mass, disruption of bone microarchitecture without alteration in the composition of bone. Osteoporotic bones are more at risk of fracture.
Note=A chromosomal aberration involving COL1A1 is found in dermatofibrosarcoma protuberans. Translocation t(17;22)(q22;q13) with PDGF. -
Sequence similarities
Belongs to the fibrillar collagen family.
Contains 1 fibrillar collagen NC1 domain.
Contains 1 VWFC domain. -
Post-translational
modificationsProline residues at the third position of the tripeptide repeating unit (G-X-Y) are hydroxylated in some or all of the chains. Proline residues at the second position of the tripeptide repeating unit (G-X-Y) are hydroxylated in some of the chains.
O-linked glycan consists of a Glc-Gal disaccharide bound to the oxygen atom of a post-translationally added hydroxyl group. -
Cellular localization
Secreted > extracellular space > extracellular matrix. - Information by UniProt
-
Database links
- Entrez Gene: 395532 Chicken
- Entrez Gene: 282187 Cow
- Entrez Gene: 282188 Cow
- Entrez Gene: 403651 Dog
- Entrez Gene: 403824 Dog
- Entrez Gene: 1277 Human
- Entrez Gene: 1278 Human
- Entrez Gene: 12842 Mouse
see all -
Alternative names
- Alpha 1 type I collagen antibody
- Alpha 2 type I collagen antibody
- alpha 2 type I procollagen antibody
see all
Images
-
Western blot - Anti-Collagen I antibody (ab233080)All lanes : Anti-Collagen I antibody (ab233080) at 2 µg/ml
Lane 1 : Human placenta lysate
Lane 2 : Human liver lysate
Lane 3 : Pig skin lysate
Secondary
All lanes : HRP-Linked Guinea pig Anti-Rabbit at 1/2000 dilution
Predicted band size: 139 kDa -
 Western blot - Anti-Collagen I antibody (ab233080)Anti-Collagen I antibody (ab233080) at 2 µg/ml + Recombinant human Collagen I protein
Western blot - Anti-Collagen I antibody (ab233080)Anti-Collagen I antibody (ab233080) at 2 µg/ml + Recombinant human Collagen I protein
Secondary
HRP-Linked Guinea pig Anti-Rabbit at 1/2000 dilution
Predicted band size: 139 kDa -
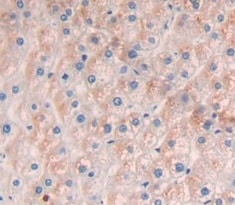 Immunohistochemistry (Formalin/PFA-fixed paraffin-embedded sections) - Anti-Collagen I antibody (ab233080)
Immunohistochemistry (Formalin/PFA-fixed paraffin-embedded sections) - Anti-Collagen I antibody (ab233080)Formalin-fixed, paraffin-embedded human kidney tissue stained for Collagen I using ab233080 at 20 µg/ml in immunohistochemical analysis. DAB staining.
Protocols
To our knowledge, customised protocols are not required for this product. Please try the standard protocols listed below and let us know how you get on.
Datasheets and documents
-
SDS download
-
Datasheet download
References (6)
ab233080 has been referenced in 6 publications.
- Zhang Z et al. Icariin regulates stem cell migration for endogenous repair of intervertebral disc degeneration by increasing the expression of chemotactic cytokines. BMC Complement Med Ther 22:63 (2022). PubMed: 35272637
- Wang T et al. Repair of osteochondral defects mediated by double-layer scaffolds with natural osteochondral-biomimetic microenvironment and interface. Mater Today Bio 14:100234 (2022). PubMed: 35308043
- Li Y et al. Three-Dimensional Microtumor Formation of Infantile Hemangioma-Derived Endothelial Cells for Mechanistic Exploration and Drug Screening. Pharmaceuticals (Basel) 15:N/A (2022). PubMed: 36422523
- Tan S et al. Fas/FasL mediates NF-?Bp65/PUMA-modulated hepatocytes apoptosis via autophagy to drive liver fibrosis. Cell Death Dis 12:474 (2021). PubMed: 33980818
- Fu XN et al. Erythropoietin enhances meniscal regeneration and prevents osteoarthritis formation in mice. Am J Transl Res 12:6464-6477 (2020). PubMed: 33194044
- Liu Y et al. Differences in microRNA-29 and Pro-fibrotic Gene Expression in Mouse and Human Hypertrophic Cardiomyopathy. Front Cardiovasc Med 6:170 (2019). PubMed: 31921893